SKU
CT14
Categories All Courses, Clinical Trials, Pharmaceutical & Biotech, Regulatory Affairs & Compliance
Clinical Trial Safety Reporting Requirements in the EU and USA
Course Summary
This course sets out the legal and regulatory requirements for safety reporting in clinical trials of medicinal products under the jurisdictions of the European Union and the USA. It builds on the foundation laid by our companion course CT13, Safety Reporting in Clinical Trials, and provides greater detail of specific requirements in those jurisdictions.
£95.00 Original price was: £95.00.£75.00Current price is: £75.00. exc. VAT
Main Menu
Back to Courses
Purchasing Information
When you have completed your course order, Zenosis will finalise the setup of your course materials and contact you on the email address that you provide in your order. You can expect this process to be completed within one business day (using the UK business calendar) of completing your payment.
It is therefore essential that you use your real email address for your order, or indicate in the purchase notes the email address to be used for the course set-up, and check that any messages from Zenosis or grapl are not lost in your junk or spam folder.
You will have access to the course module(s) for a period of 180 days after your purchase is complete.
Detailed Course Information
Learning Objectives
• Identify relevant EU statutes and sources of regulatory guidance
• Identify online portals that are key to safety reporting in clinical trials in the EU
• Summarize investigators’ and sponsors’ responsibilities under the Clinical Trials Regulation
• Discuss the role of reference safety information in the EU
• Specify sponsors’ responsibilities for reporting suspected unexpected serious adverse reactions in the EU
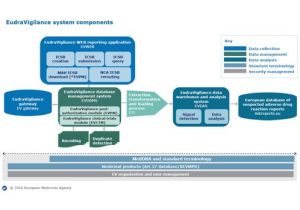
• Describe how to submit electronic reports to EudraVigilance
• Outline sponsors’ responsibilities for reporting SUSARs to investigators under the Clinical Trials Regulation
• Identify submissions that sponsors must make to the EU Clinical Trials Information System
• Outline significant differences in requirements under the Clinical Trials Directive
• Identify relevant US statutes and sources of regulatory guidance
• Summarize clinical investigators’ responsibilities for reporting to sponsors of trials conducted under an Investigational New Drug application (IND) to the US Food and Drug Administration (FDA)
• Discuss the assessment of causality of serious adverse events
• Summarize sponsors’ responsibilities for review of safety information under an IND
• Specify sponsors’ responsibilities for IND safety reporting to FDA and investigators
• Describe how to deal with anticipated events according to FDA guidance
• Specify timeframes for IND safety reporting
• Specify requirements for analysis of similar events and submission of follow-up information
• Describe how to submit IND safety reports to the FDA
• Discuss requirements for electronic submission of IND safety reports
• Discuss requirements for investigators’ reporting of unanticipated problems to investigational review boards
• Specify sponsors’ responsibilities for submission of IND annual reports
Who will benefit from this course
This course provides essential information for clinical research, drug safety, and regulatory affairs staff of sponsors of clinical trials, as well as investigators and other healthcare professionals who undertake clinical trials.
Module Outline
• Legal and regulatory requirements in the EU
– In this session, we set out the legal and regulatory requirements for safety reporting under the EU Clinical Trials Regulation. We specify the responsibilities of investigators and those of sponsors. We distinguish those reports that must be submitted by sponsors to the EudraVigilance portal and those that must be submitted to the Clinical Trials Information System. We specify the format and terminology that must now be used, and we identify the tools and pathways for electronic submission. Finally, we outline significant differences in requirements under the Clinical Trials Directive.
• Legal and regulatory requirements in the USA
– In this session, we set out the legal and regulatory requirements for safety reporting in clinical trials conducted (in the USA or elsewhere) under an Investigational New Drug application (IND) to the US Food and Drug Administration (FDA). We specify the responsibilities of investigators and those of sponsors. We describe the criteria for IND safety reports to the FDA, and the content, format and timing of their submission. We discuss investigators’ obligation to report unanticipated problems to institutional review boards. Finally, we discuss IND annual reports and other safety reporting issues.
• Assessment
– Multiple-choice mastery assessment.
Roles
Clinical research, drug safety, and regulatory affairs staff, healthcare professionals
CPD Points
2
Region
Level
Intermediate, Advanced
Course Study Time
2 hours
Europe, USA